


2025-03-07

临床试验需保证参与受试者不会承担可以避免的安全性风险,也需保证试验持续足够的时间,不会因过早终止而不能回答预设的科学问题。另外针对设置有期中分析任务的盲态确证性临床试验,期中分析时需根据非盲态数据进行分析(如PFS、OS等终点),判断干预措施是否达到预设的优效性或无效性边界,若疗效显著,可提前终止试验以加速药物上市;若疗效不足,则可能需要终止以避免资源浪费,针对是否终止试验的建议若由申办方或研究者主导,可能会因为利益冲突等原因导致试验结果的可信度。因此,临床试验有时就需要成立独立的临床试验数据监查委员会(Independent Data Monitoring Committee, IDMC)来承担这些任务。那么什么是IDMC呢?它的目的和意义何在?项目中如何实施呢?……今天让我们一起来揭秘!
IDMC的起源
IDMC简称临床试验数据监查委员会(Data Monitoring Committee,DMC),又称数据安全监查委员会(Data and Safety Monitoring Board, DSMB),它是一个独立的具有相关专业知识和经验的专家组,负责定期审阅来自一项或多项正在开展的临床试验的累积数据,从而保护受试者的安全性、保证试验的可靠性以及试验结果的有效性。为了行文方便,以下除非特别说明,统一用IDMC代表独立临床试验数据监查委员会或数据安全监查委员会。
它的起源可以追溯到20世纪60年代初,最初主要用于由美国联邦机构资助的大规模多中心随机试验,例如美国国立卫生研究院(NIH)和退伍军人事务部(VA)等机构发起的试验。1967年,NIH的一个外部咨询团体首次引入了正式的委员会概念,负责在试验过程中审查不断积累的数据,以监测安全性、有效性和试验执行问题,并向当时的国家心脏研究所提出建议。这一建议的提出是基于对期中监测的必要性认识,即确保参与试验的受试者的安全性,同时避免与试验设计和实施密切相关的人员因利益冲突而无法客观评估数据。此后,IDMC在全球范围内的应用逐渐广泛。2005年欧洲药品管理局(European Medicines Agency,EMA)发布了IDMC指南《Guidance on data monitoring committees》,2006年,美国食品药品监督管理局(Food and Drug Administration,FDA)发布了关于IDMC的指导原则《Guidance for clinical trial sponsors, establishment and operation of clinical trial data monitoring committees》。2020版《药物临床试验质量管理规范》中,第五章第三十六条(二)提到申办者可以建立IDMC,以定期评价临床试验的进展情况,包括安全性数据和重要的有效性终点数据,IDMC可以建议申办者是否可以继续实施、修改或者停止正在实施的临床试验,独立的数据监查委员会应当有书面的工作流程,应当保存所有相关会议记录。为指导临床试验申办者建立药物临床试验IDMC,规范IDMC的监查活动,2020年9月中国国家药品监督管理局(National Medical Products Administration,NMPA)药品审评中心(CDE)发布了《药物临床试验数据监查委员会指导原则》,进一步规范了iDMC的建立与运行。
IDMC的目的和意义
IDMC在临床试验中扮演关键角色,其核心目的是通过独立、客观的定期审阅来自一项或多项正在开展的临床试验的累积数据确保试验的安全性、可靠性、有效性以及伦理合规性,其详细目的和意义总结如下:
安全性监查
IDMC的首要任务是进行安全性监查以保护受试者的安全,定期监查试验中积累的安全性数据(如不良事件发生率)。监查过程中,如果发现重大安全性问题、或者对安全性问题存在严重担忧,IDMC可能会考虑向申办者提供终止临床试验、暂停试验并进一步查明试验的安全性问题等建议。
在试验开始前,申办者应与IDMC成员充分讨论试验中可能观察到的所有值得特别关注的潜在不良事件和不良反应。即便如此,在安全性监查时仍可能遇到一些事先未曾考虑到的情况,比如其它已完成或正在进行的相关临床试验发布的外部安全性信息,对此IDMC需要了解更多的细节和额外信息,才能做出正确判断。
有效性监查
IDMC的一个重要任务是通过审阅期中分析数据对有效性进行监查,并协助申办者做出是否提前终止试验的决策以节省资源或避免受试者继续接受无效治疗。提前终止试验的建议主要包括以下两种情况:
试验操作质量监查
IDMC还可以通过审阅试验数据对试验操作质量进行监查,包括监查方案依从性、招募状态、受试者的脱落率和数据完整性等方面的信息。如果发现试验执行过程中出现严重质量问题,IDMC 应建议申办者改善研究质量。例如,IDMC通过审阅对所收集数据的分析结果,发现随机化错误、缺失数据比例太大或组间基线严重不均衡等问题,有必要及时建议申办者找出产生问题的原因并加以解决。
试验设计调整
在试验设计中若初始样本量估算不足,IDMC可基于期中分析结果重新计算样本量,确保试验的统计学把握度。对于采用适应性设计等复杂设计类型的临床试验,常需要基于已收集数据,对正在进行的试验要素进行调整和修改,如干预剂量、研究人群,或用于样本量估计的效应量及误差等。此时作为独立第三方的IDMC的参与是非常必要的。可以由IDMC根据事先在研究方案及IDMC章程中明确规定的规则,在保证试验完整性的前提下,对正在进行的试验设计提出调整的建议,这将有助于提升试验的科学性,并降低试验失败的风险。
IDMC应执行研究方案中预设的计划,而不应直接参与研究方案的修订,特别是与有效性评价相关的方案修订。当涉及根据外部数据对试验设计调整时,也应由申办者,而不是IDMC,提出试验设计调整(如调整终点指标、改变或增加预设亚组等)。
值得注意的是IDMC的主要作用是提供建议,而其建议是否被接受则由申办者决定。
临床试验中是否需要设立IDMC,需根据研究项目的具体需求而定。例如,大多数早期探索性试验、没有重大安全性问题的短期研究,可能不需要设立专门的 IDMC;而确证性临床试验,特别是大样本、安全性风险高、包含适应性特征的复杂设计,或者观察周期较长的临床试验,设立IDMC就显得非常必要。即使是开放性试验,包括单臂试验,若有必要在试验过程中评估汇总数据,申办者也应考虑设立IDMC,需要IDMC的试验可总结如下:
IDMC的流程
IDMC流程的核心阶段包括前期准备工作、IDMC会议阶段以及申办方决策与后续行动,前期准备工作包括IDMC组员的成立、IDMC章程的撰写、IDMC SAP和TFL shell的绘制、统计分析程序的创建等,IDMC会议包括开门会议和闭门会议,闭门会议结果形成书面建议,提交给申办方决策者。
期中分析是IDMC最常见的应用场景,针对此类项目,基于各职能部门绘制了IDMC详细执行流程,参见以下图示。
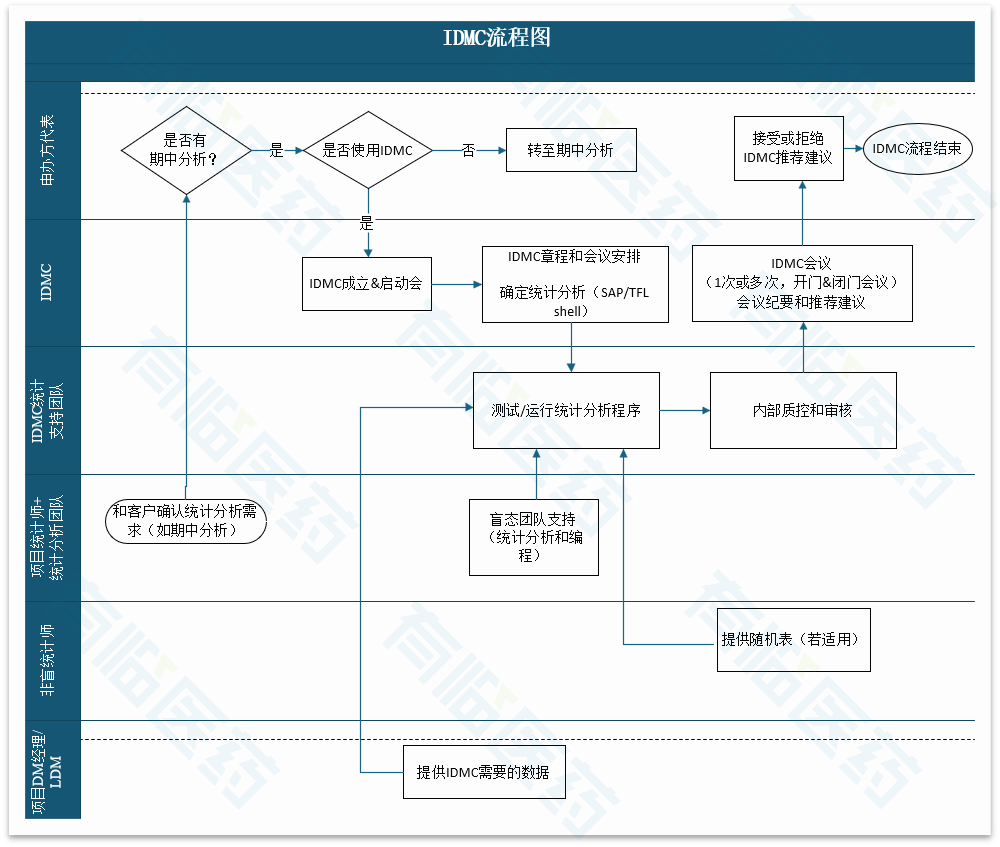
案例分享和总结
有临数统团队在过去几年间,承接多项临床试验IDMC独立统计支持工作,包括部分早期以及多项II/III期研究。并在更多研究里积累了作为盲态团队与IDMC以及独立统计支持团队(iSSG)合作的经验。
接下来以近期执行的一项IDMC试验为基础模拟一个示例,试验为随机对照双盲多中心II/III期临床研究,其大致历程可参见以下图示。
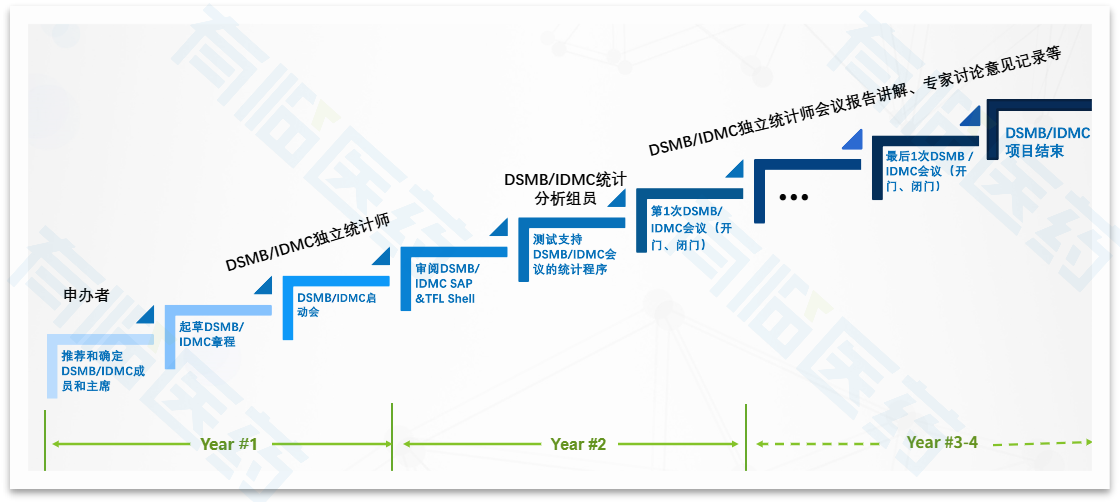
根据既往项目经验,在执行IDMC时,需特别注意以下关键事项,以确保受试者安全、数据完整性和试验科学性。
独立性维护
避免利益冲突
隔离非盲团队
数据质量管理
数据及时性与完整性
揭盲流程控制
统计分析与风险控制
多重性校正
期中分析设计
统计分析编程
沟通与决策机制
明确沟通层级
应急管理
盲态保持
多方进行沟通时,无论邮件、会议或其他方式,均不得讨论、展示或暗示盲底信息,即使非盲成员间沟通时,亦不可公开讨论、发送带有盲底的图片等。
伦理与法规合规
脆弱人群保护
区域法规差异
多区域试验(MRCT)需考虑不同地区监管要求(如FDA、EMA、NMPA对IDMC章程的差异),必要时设立区域子委员会协调审查。
文档与记录
全程留痕
数据溯源
非盲统计图表需标注版本号和生成时间,确保与盲态数据的一致性。
存档
有关会议纪要、建议书等文档定稿后需尽快存档,并由专人保管,避免遗失等。
挑战与应对策略
挑战1:数据不一致
应对:提前协调研究者与IRC的评估标准(如RECIST 1.1),统一培训以减少分歧。
挑战2:揭盲风险
应对:采用独立数据监查平台,自动化生成非盲报告,减少人工干预。
挑战3:统计复杂性
应对:引入外部统计顾问审核分析计划,确保方法符合ICH E9(R1)指导原则。
简言之,执行IDMC的核心在于通过独立性、透明度和规范性平衡科学探索与伦理责任,从而达到保障临床试验的科学性与完整性、及时终止高风险或无效试验、提前终止有效性试验以加速药物上市等目的。
参考文献:
[1]《药物临床试验数据监查委员会指导原则(试行)》,国家食品药品监督管理总局2021年第63号
[2]《药物临床试验质量管理规范(2020版)》,国家药品监督管理局2020年第57号
[3]Establishment and Operation of Clinical Trial Data Monitoring Committees. U.S. Department of Health and Human Services Food and Drug Administration. 2006.3
[4]Guideline On Data Monitoring Committees, Committee For Medicinal Products For Human Use. European Medicines Agency. 2005.7
撰写:李纪杰、何志豪
审核:章飞燕、张子豹










