


2026-06-18
引言:
一份面向申请人的操作手册
国家药监局药审中心与 5 月 28 日发布《罕见疾病创新药物研发鼓励试点计划(“关爱计划-延伸”)》,对于有志于投身罕见病药物研发的创新药企而言,这不仅是一项政策利好,更是一份清晰的操作指南。该计划旨在通过“早期介入、一品一策”的工作模式,帮助企业在研发早期与监管机构建立紧密的沟通桥梁。
本文从申请人(企业)的角度出发,聚焦如何申请、需要准备什么材料、纳入后如何执行等关键操作性问题,对《申报指南》和《罕见疾病创新药物临床研发实施框架》进行深入剖析,帮助企业更好理解和运用这一计划。
01
如何判断你是否符合申请资格?
在开始准备申请材料之前,企业首先需要自我审视:我的品种是否符合“关爱计划-延伸”的准入门槛?根据《申报指南》1.1节,申请品种必须同时满足以下三个条件,缺一不可:
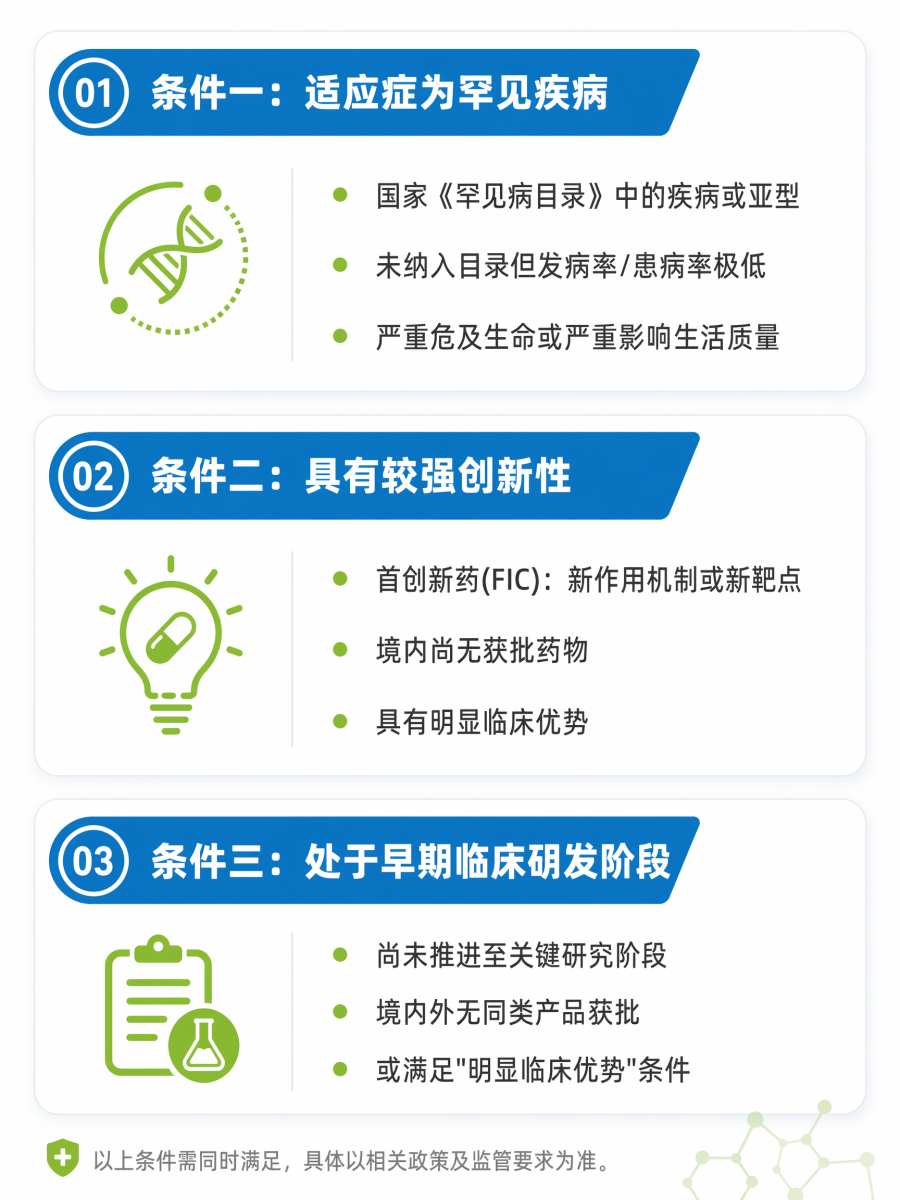
02
申请实操详解:流程与材料清单
一、申请方式与流程(《申报指南》1.3 节)
申请遵循 “自愿申请、沟通交流” 的原则,具体流程如下:
1.递交申请:申请人通过药审中心"申请人之窗",选择沟通交流途径,提交加入"关爱计划-延伸"试点工作的申请。
2.明确备注:在沟通交流申请的"会议目的"和"其他情况说明"两项中,必须备注填写"关爱计划-延伸申请"。这是一个非常重要的操作细节,便于药审中心快速识别并将其归入正确的审评通道。
3.独立申请:此次沟通交流不建议与其他事项合并(如研发技术问题咨询),应专注于"关爱计划-延伸"的申请本身。
4.审评与公示:药审中心审评团队对申请进行评价,必要时召开沟通交流会。经评价拟同意的品种,将在药审中心网站进行为期5个工作日的公示。公示内容包括药品名称、申请单位、拟开发的适应症。无异议后,品种正式纳入试点。
二、申请资料详解——
核心在于《实施框架》(《申报指南》1.4 节)
申请所需的核心材料,除了《药审中心沟通交流管理办法》要求的一般材料外,最关键、最核心的是必须填写并提交《罕见疾病创新药物临床研发实施框架》。
这份《实施框架》是申请的重中之重,它既是您向药审中心展示研发思路的蓝本,也是品种纳入后动态管理的档案。以下是《实施框架》各章节的操作性填写指南:包含七个章节,核心回答“是什么、为什么、怎么做、如何跟踪”四个问题。
章节一:目标适应症诊疗情况与患者参与计划
这是回答"我们为什么要治这个病"以及"我们对疾病有多了解"。
1.当前诊断、治疗及未满足需求:需清晰总结疾病特征(病因、发病人群、临床表现、预后、患病率/发病率)、现有诊断方法、治疗策略,并明确指出目前未被满足的临床需求是什么。
2.自然病史研究:这是医药研发的基石。需说明是否已开展或在计划中开展。特别重要的是:如果自然病史研究数据未包含中国患者,必须详细评估并说明其对中国的相关性。强烈鼓励在中国开展自然病史研究,这可直接用于优化临床试验设计或作为外部对照数据评价疗效。
3. 患者参与计划:强调从患者视角获取体验数据。需阐述如何收集患者观点、诊治现状、未满足需求、治疗获益期望等。这些数据有助于明确临床需求,为临床试验设计提供患者视角的依据。
章节二:产品概况与立题依据
这是回答"你的药凭什么能治这个病"。
需结合立题依据和药品作用机制(包括潜在的毒性/危害、药效学相互作用),对药物的安全性和有效性提出合理预期。重点说明该产品预期能够填补哪些未满足的临床需求,或比现有治疗有何优势。这是决定审评团队是否认可"立题合理"的关键部分。
章节三:目标适应症临床开发计划
这是回答"你打算怎么证明你的药有效又安全"。这是整个《实施框架》中最核心的战略部分。
1.临床开发计划概要及注册策略:需清晰概述整体研发计划,包括已开展的试验进展、未来研究安排、是否采用全球多中心研究等。尤其要强调如何利用外部数据(如自然病史数据)进行设计优化。需要提供一个清晰的总结表,列明所有计划中、进行中、已完成的研究,并说明它们之间的相互关系与先后顺序。
2.临床结局评估(COA)开发计划:如果该病种没有成熟的疗效评价终点,鼓励开发新的患者报告结局(PRO)、观察者报告结局(ObsRO)或临床医生报告结局(ClinRO)等COA工具。
3.初拟目标产品特征(TPP)概要:TPP是说明书的雏形。需按照说明书的框架,写出预期的产品特征,如适应症、用法用量、禁忌症、不良反应等。
章节四至七:细节验证与沟通计划
四、已开展/拟开展的临床试验设计及结果:对于每项试验,都需详细说明研究设计、目的、人群、剂量、样本量依据。如有结果需概述。这为审评团队提供了验证战略是否可行的具体证据。
五、六、非临床/药学计划:简要介绍非临床和药学的开发计划,重点在于其与临床开发计划的匹配性,即如何支撑目标适应症的上市申请。
七、与药审中心沟通交流计划:申请时无需填写此章。纳入试点后,将由药审中心与企业共同商定初步的里程碑式沟通计划,重点包括Pre-IND、EOP1、EOP2、Pre-NDA等关键节点的沟通,以及每次沟通后的总结纪要。

03
纳入试点后的项目管理与执行
一.纳入后的权利与义务(《申报指南》二、项目的执行)
要点一
管理路径不变:纳入试点的品种,其常规的申报、审评、注册工作(如IND、NDA),仍需严格依据现行法规和工作程序执行。
要点二
动态更新与主动报告:企业需根据研发进展,不断更新《实施框架》。当发生"重大研发方向变更"(如适应症调整、关键试验方案重大修改)或因安全性事件导致开发暂停/终止时,必须及时向药审中心书面报告。
要点三
阶段性汇报:企业还需根据审评需求,阶段性汇报相关工作进展或结果。这并非增加负担,而是确保信息畅通、指导精准的重要手段。
二、核心优势与“温馨提示”
准备要充分:申请前务必仔细研读《申报指南》和实施框架清单,确保资料完整、逻辑自洽。自然病史研究、患者体验数据等需提前布局
沟通要主动:不要等到问题出现才去沟通。主动、定期地与审评团队保持联系,利用好“里程碑式沟通计划”和“日常沟通交流”渠道。
框架要动态:《实施框架》不是一成不变的,要根据研发数据和外部环境的变化及时更新。每次更新都应在系统内有清晰的版本记录。
法规要遵循:试点计划是锦上添花,不是雪中送炭。所有操作必须以遵守《药品管理法》等现行法律法规为前提。
结语:
主动拥抱监管,加速创新转化
“关爱计划-延伸”是药审中心送给罕见病创新药企业的一份“厚礼”。它不仅降低早期研发的不确定性,更开启一种全新的、协同式的研发监管模式。对于企业来说,关键在于主动学习、精心准备、动态管理。
将《实施框架》视为一份与监管机构共同绘制的研发蓝图,将每一次沟通视为寻找方向、规避风险的宝贵机会。只有这样,才能真正用好这一政策红利,将源头创新的火花,更快、更稳地转化为惠及罕见病患者的良药。











