


2026-02-27
有临医药
随着生活方式的改变以及人口老龄化的加剧,高尿酸血症与痛风已然成为全球性的公共健康难题。作为肾脏尿酸重吸收的关键调控蛋白,尿酸转运蛋白1(URAT1)成为了痛风治疗的“总闸门”靶点。本文系统地梳理了URAT1抑制剂的研发历程,从早期的非选择性药物发展到新一代的高选择性抑制剂,揭示了提高分子选择性对于规避非靶点毒性的核心重要性。通过整合近期的结构生物学、临床前以及临床研究数据,深入剖析了URAT1抑制剂的分子作用机制、关键临床评价维度(如分子选择性、药代动力学特征以及深层组织疗效)以及当前全球的研发格局。最后,展望了该领域朝着精准医疗与联合治疗方向发展的未来趋势,强调了解析URAT1结构与功能关系对于设计更安全有效的药物具有指导价值。
一、引言:高尿酸血症与痛风的疾病负担与治疗挑战
高尿酸血症指的是血液中尿酸水平持续升高,通常将血清尿酸(sUA)> 7.0 mg/dL定义为高尿酸血症[1]。长期高尿酸血症是痛风发生的直接原因,当sUA超过饱和点,尿酸盐结晶析出并沉积于关节及周围组织,引发剧烈炎症反应,以及急性痛风性关节炎。若未得到有效控制,疾病将进展为慢性痛风石性痛风,导致关节破坏、功能丧失和慢性疼痛[2]。全球范围内,痛风的患病率正逐年上升,给患者生活质量和社会医疗体系带来沉重负担[3]。
尿酸是人类嘌呤代谢的终产物。人体内尿酸水平的稳态由生成与排泄之间的平衡所决定。大约三分之二的尿酸通过肾脏排泄,其余三分之一则经肠道排出[4]。在肾脏,肾小球自由滤过的尿酸约90%在近曲小管被重吸收回血液循环,仅约10%最终随尿液排出。因此,肾脏尿酸排泄不足是绝大多数(约90%)高尿酸血症患者的主要病因[5]。负责这一重吸收过程的关键转运蛋白便是URAT1(由SLC22A12基因编码),它位于肾近曲小管上皮细胞的顶膜(管腔侧),通过以尿酸盐交换细胞内单羧酸阴离子(如乳酸、丙酮酸)的方式工作[6]。抑制URAT1的功能,即可有效阻断尿酸重吸收,增加尿酸排泄分数,从而降低血尿酸水平。
然而,URAT1抑制剂的研发之路并非一帆风顺。从早期的丙磺舒、苯溴马隆,到近期获批的雷西纳德、多替诺雷,药物的安全性问题(尤其是肝毒性和肾毒性)始终是临床应用的制约因素。这些教训深刻地揭示了非选择性抑制其他关键转运体(如OAT1、OAT3)或药物代谢产物毒性所带来的风险。随着对尿酸转运系统认识的不断深入以及结构生物学技术的突破,新一代高选择性、高安全性的URAT1抑制剂正不断涌现。本文旨在从分子机制、临床研发与审评的视角,系统地阐述URAT1抑制剂的“前世今生”,并对其未来发展进行展望。
二、URAT1的生理功能与作为治疗靶点的确立
URAT1是维持血尿酸水平的关键分子。遗传学研究为此提供了最为有力的证据:SLC22A12基因的功能缺失性突变会导致遗传性肾性低尿酸血症,患者的血尿酸水平极低,且几乎不会发生痛风[7]。这反向证明了URAT1功能正常是高尿酸血症存在的“必要条件”。
尿酸的处理是一个多步骤、多转运体参与的过程(图1)。肾小球滤过的尿酸进入近曲小管管腔。位于顶膜的URAT1将管腔中的尿酸转运入细胞内,同时将细胞内的单羧酸阴离子(如乳酸)交换至管腔。随后,位于基底膜的葡萄糖转运蛋白9(GLUT9,由SLC2A9基因编码)负责将细胞内的尿酸转运至肾间质,最终回流入血[8]。此外,位于基底膜的有机阴离子转运蛋白1和3(OAT1/OAT3)介导了尿酸从血液向近曲小管细胞内的摄取,这一步骤是尿酸最终经顶膜分泌泵(如MRP4、NPT4)排入管腔的前提[9]。URAT1因其承担了绝大部分(约90%)滤过尿酸的重吸收任务,而成为促尿酸排泄治疗的“阿喀琉斯之踵”。
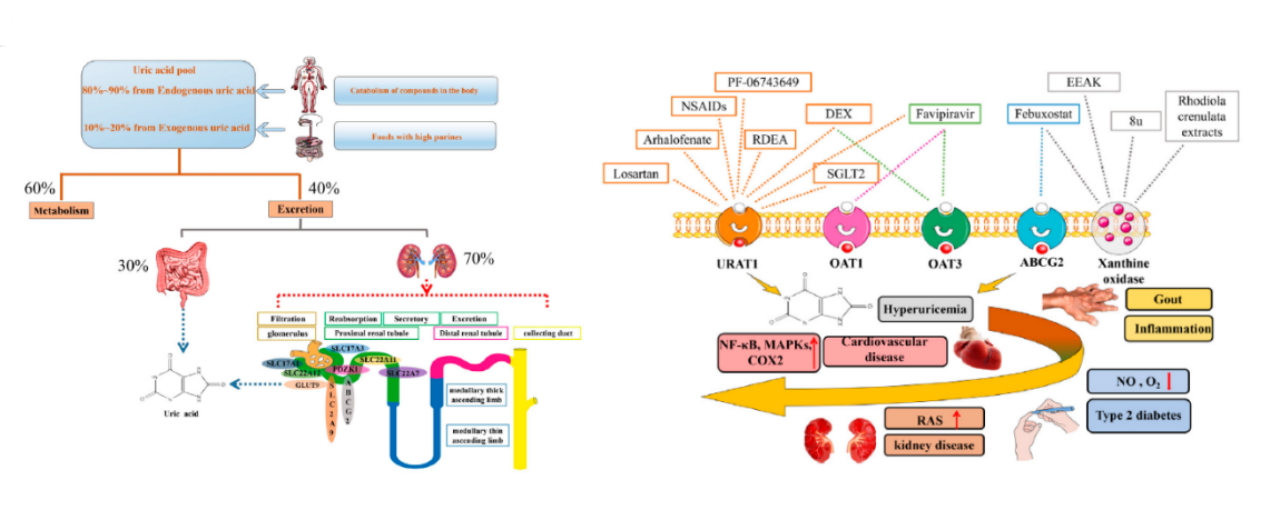
图1 体内尿酸的生理变化机制
2. 高尿酸血症与多种疾病的关联
越来越多的证据显示,高尿酸血症不仅是痛风的病因,还与多种慢性疾病的发病和预后密切相关,包括慢性肾脏病(CKD)、心血管疾病(高血压、冠心病、心力衰竭)、代谢综合征(胰岛素抵抗、肥胖、脂代谢紊乱)等[10]。其机制涉及氧化应激、内皮功能障碍、肾素-血管紧张素系统激活以及慢性炎症等。因此,有效控制血尿酸水平具有超越痛风治疗本身的更广泛临床意义。
三、URAT1抑制剂的研发谱系:从非选择性到高选择性的进化
URAT1抑制剂的研发历史,是一部对“选择性”的理解不断深化、对分子结构不断优化的历史。根据其选择性与研发时代,大致可分为三代。
第一代:非选择性抑制剂——丙磺舒(Probenecid)
丙磺舒于1949年合成,最初用于延缓青霉素的肾脏排泄。随后发现它具有促尿酸排泄的作用。然而,丙磺舒对URAT1的选择性极差,它同时强力抑制OAT1和OAT3[11]。OAT1和OAT3是肾脏清除多种内源性物质(如前列腺素)和外源性药物(如甲氨蝶呤、抗生素、利尿剂)的关键转运体。因此,丙磺舒的使用伴随着显著的药物相互作用风险,限制了其临床应用。此外,其降尿酸效力相对温和,在别嘌醇问世后,其使用逐渐减少。
第二代:效力增强但安全性存疑——苯溴马隆(Benzbromarone)
苯溴马隆于20世纪70年代上市,其对URAT1的抑制效力达到纳摩尔级别,降尿酸效果显著,一度在欧洲和亚洲广泛应用。然而,2003年因其引起的严重、甚至致死性肝毒性病例报告而在欧洲撤市[12]。其肝毒性机制与细胞色素P450 2C9(CYP2C9)代谢产生的活性中间体(如6-羟基苯溴马隆)有关,这些代谢产物可引起线粒体损伤和免疫介导的肝细胞坏死[13]。尽管在中国、日本等地,苯溴马隆在严格肝功能监测下仍在使用,且对肾功能不全患者相对安全(因其主要经肝代谢),但肝毒性阴影始终笼罩其上。苯溴马隆同样存在非选择性问题,对OAT1、OAT3及GLUT9也有抑制作用[14]。
第三代:高选择性抑制剂的崛起
新一代URAT1抑制剂的核心设计理念是“高选择性”,旨在最大限度地抑制URAT1,同时避免影响OAT1/OAT3等其他关键转运体,从而从源头上降低药物相互作用和肾小管毒性风险。
四、结构生物学突破:揭示URAT1抑制剂的分子作用机制
长期以来,URAT1抑制剂如何与靶点结合、其作用模式是怎样的,一直是个未解之谜。2025年,Suo等人发表在《自然-通讯》(Nature Communications)上的研究,利用冷冻电镜技术首次解析了URAT1与三种抑制剂(苯溴马隆、雷西纳德和新型化合物TD-3)复合物的高分辨率结构,为我们理解URAT1抑制机制带来了革命性的见解[18]。
1.URAT1的结构与抑制剂结合口袋
URAT1属于主要协助超家族转运蛋白,具有典型的“倒V字”型结构,包含12个跨膜螺旋。研究揭示,无论是否结合抑制剂,URAT1均稳定在向内开放的构象。抑制剂结合在由跨膜螺旋形成的中央结合口袋中。该口袋可划分为几个关键区域:
2.非竞争性抑制模式
功能性实验结合结构分析发现,苯溴马隆、雷西纳德和TD-3均为URAT1的非竞争性抑制剂。这与多数转运蛋白抑制剂通常以竞争性方式结合于向外开放构象不同。研究者提出,由于URAT1表达在肾小管细胞的顶膜(朝向管腔),抑制剂可能首先进入细胞,再从胞内一侧结合稳定URAT1的向内开放构象,从而阻断其转运循环[18]。这一机制解释了为何抑制剂倾向于结合向内开放状态,以及为何它们与底物尿酸并非直接竞争同一结合位点。
3.不同抑制剂的结合特征
这些结构信息为理性设计下一代具有更高选择性、更强效力和更佳安全性的URAT1抑制剂提供了宝贵的蓝图。
五、临床审评的核心维度:超越降尿酸效力的综合评价
对于一款URAT1抑制剂的临床价值评估,现代审评视角已从单一的降尿酸幅度,转变为一个多维度的综合评价体系。
1.分子选择性:安全性的基石
不仅要关注对URAT1的半抑制浓度(IC50),更要评估其对相关转运体的选择性倍数。
表 1 不同URAT1抑制剂的关键转运体的体外抑制效力(IC50,微摩尔)
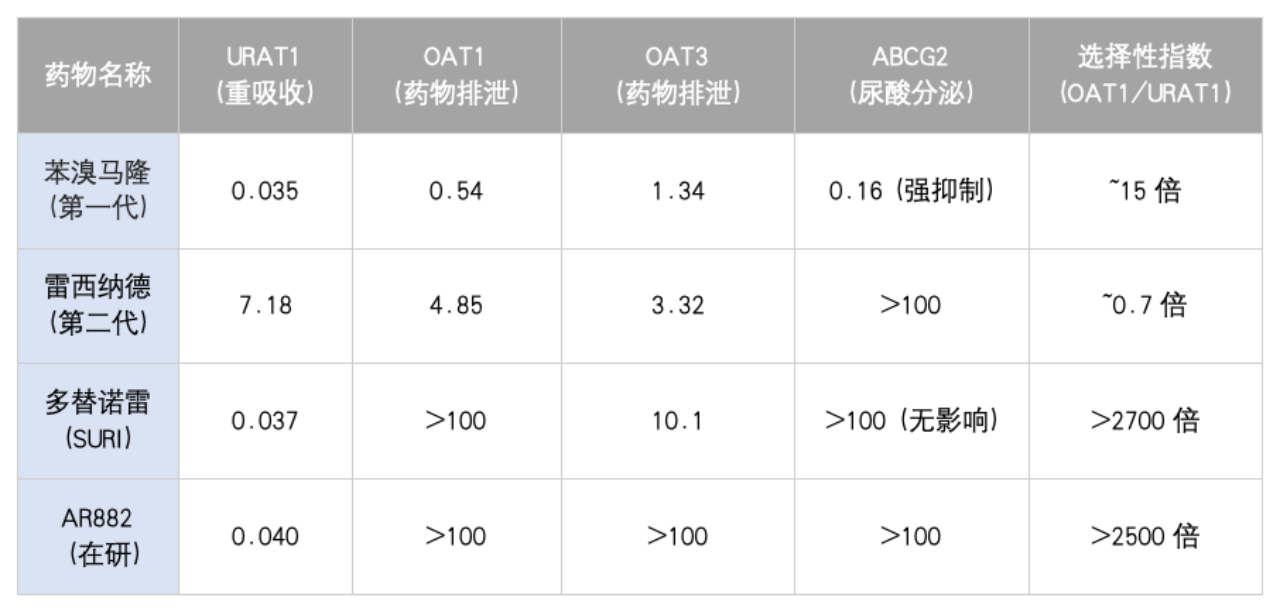
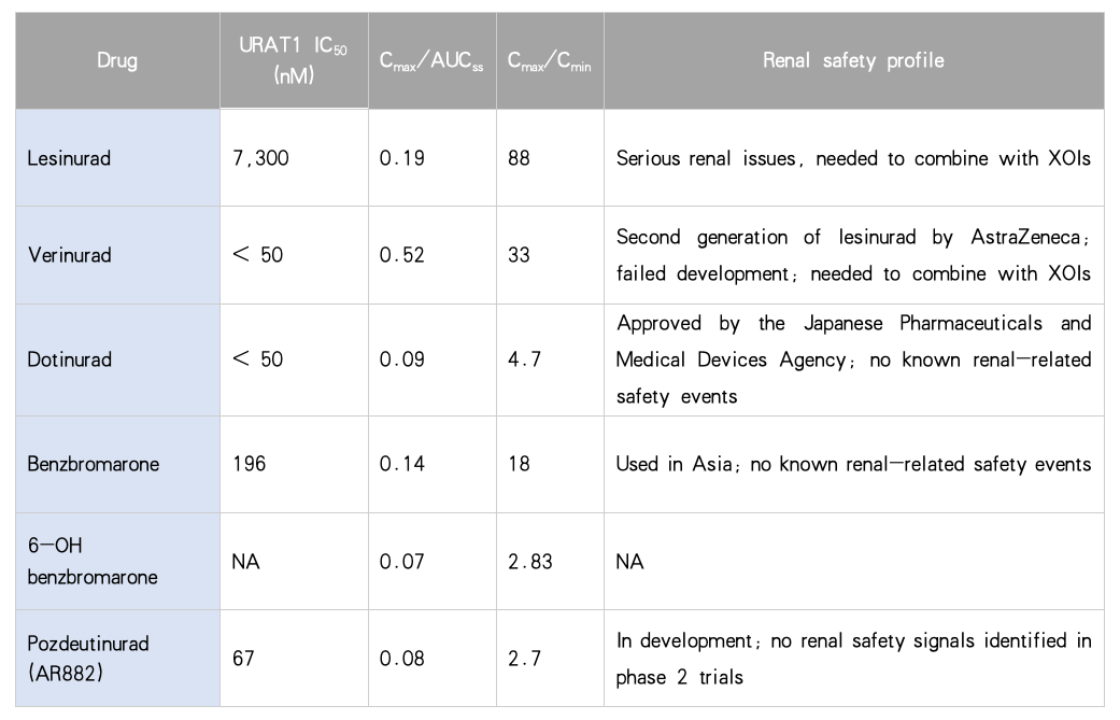
近年研究发现,稳态时最大血药浓度与药时曲线下面积的比值(Cmax/AUCss)是一个预测肾脏安全性的潜在指标[20]。较高的比值意味着药物在体内形成尖锐的血药浓度峰,可能导致短时间内大量尿酸涌入肾小管,超过其溶解度,形成微结晶,从而诱发一过性肾小管损伤和血清肌酐升高。雷西纳德和维瑞纳德(Verinurad)的临床研究都曾观察到与剂量相关的肾脏不良事件。新一代抑制剂如多替诺雷和AR882通过优化分子结构和剂型,实现了更平缓的药物暴露,这可能是其肾脏安全性更佳的重要原因。
表 2 不同URAT1抑制剂稳态Cmax/AUCss和Cmax/Cmin 比值[21]
3.疗效的纵深:从血尿酸达标到痛风石溶解
对于慢性痛风石患者,治疗的最终目标是溶解已形成的尿酸盐结晶,逆转疾病。因此,评估URAT1抑制剂的疗效不能仅停留在血尿酸达标率。
六、全球及中国研发格局与未来展望
1.当前研发梯队
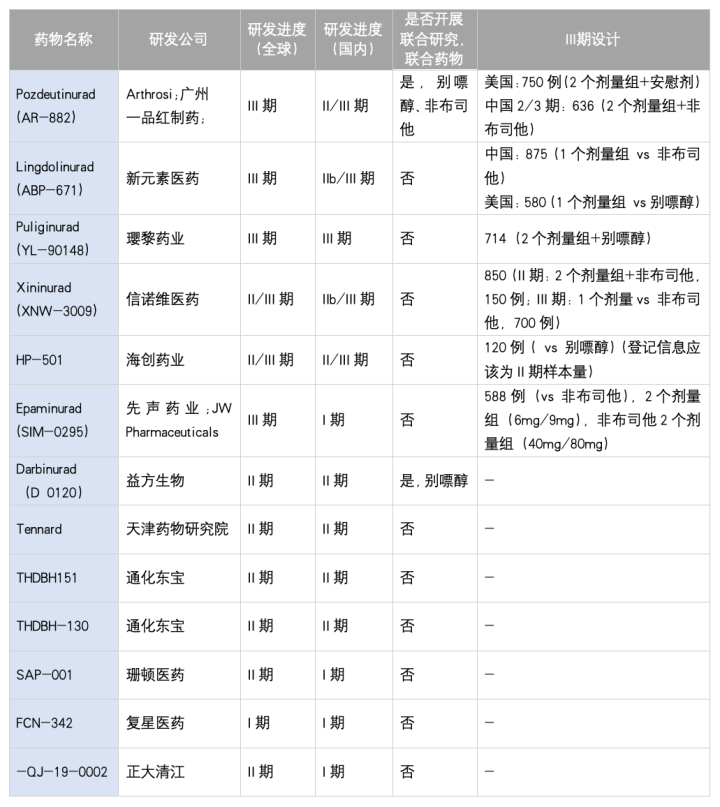
当前URAT1抑制剂的研发已进入密集收获期,形成了传统药物、已上市新药和在研新药并存的格局(表3)。
表 3 中国URAT1抑制剂临床研究开展情况
2.未来发展方向
七、总结
URAT1抑制剂的演进历程,是现代药物研发从粗放到精准、从经验到理性的一个缩影。从丙磺舒的广泛抑制,到苯溴马隆的效力与毒性并存,再到新一代药物对URAT1选择性的极致追求,每一步都伴随着对疾病生物学和药物化学理解的深化。冷冻电镜结构生物学的突破,首次让我们在原子层面“看见”了药物与靶点的相互作用,为基于结构的药物设计打开了大门。
临床审评的核心也已从单纯的疗效考核,演变为对分子选择性、药代动力学特性、深层组织疗效以及长期安全性的系统性评估。一款成功的URAT1抑制剂,必须在效力、选择性、安全性以及患者用药便利性之间取得最佳平衡。
随着多替诺雷在中国获批,以及SHR4640、AR882等一批国产创新药即将步入市场,中国痛风治疗领域正迎来“源头创新”带来的治疗升级。我们期待这些更安全、更有效的药物能够惠及广大患者,最终实现控制“疼痛之王”、改善患者长期预后的目标。未来,结合基因分型的精准治疗和多靶点联合策略,有望为痛风及高尿酸血症相关疾病的管理带来新的变革。
参考文献
[1] 徐东,朱小霞,邹和建,等. 痛风诊疗规范. 中华内科杂志,2023,62(09):1068-1076.
[2] Richette P, et al. 2016 updated EULAR evidence-based recommendations for the management of gout. Ann Rheum Dis. 2017.
[3] Sun H, et al. Function of Uric Acid Transporters and Their Inhibitors in Hyperuricaemia. Front Pharmacol. 2021.
[4] Jessica Maiuolo J, et al. Regulation of uric acid metabolism and excretion. Int J Cardiol. 2016.
[5] Enomoto A, et al. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature. 2002.
[6] Hosoyamada M, et al. Function and localization of urate transporter 1 in mouse kidney. J Am Soc Nephrol. 2004.
[7] Ichida K, et al. Clinical and molecular analysis of patients with renal hypouricemia in Japan-influence of URAT1 gene on urinary urate excretion. J Am Soc Nephrol. 2004.
[8] Vitart V, et al. SLC2A9 is a newly identified urate transporter influencing serum urate concentration, urate excretion and gout. Nat Genet. 2008.
[9] Jutabha P, et al. Human sodium phosphate transporter 4 (hNPT4/SLC17A3) as a common renal secretory pathway for drugs and urate. J Biol Chem. 2010.
[10] 孙浩路等. 尿酸转运蛋白及其抑制剂在高尿酸血症中的功能. 药理学前沿. 2021.
[11] Keenan RT, et al. How URAT1 inhibitors can shape the future of chronic gout treatment. Explor Musculoskelet Dis. 2024.
[12] Lee MH, et al. The liver toxicity of benzbromarone: a case report and review of the literature. J Clin Gastroenterol. 2003.
[13] Arai M, et al. Involvement of metabolism in benzbromarone-induced hepatotoxicity. Drug Metab Dispos. 2012.
[14] Receptors inhibited by non-selective and selective uricosurics. In: Keenan RT, et al. Explor Musculoskelet Dis. 2024.
[15] Bardin T, et al. Lesinurad in combination with allopurinol: results of a phase III study in gout patients with an inadequate response to allopurinol. Ann Rheum Dis. 2017.
[16] Shen Z, et al. Pharmacokinetics, pharmacodynamics, and safety of lesinurad, a selective uric acid reabsorption inhibitor, in healthy adult male volunteers. Clin Drug Investig. 2015.
[17] Hosoya T, et al. Dotinurad: a novel selective urate reabsorption inhibitor for the treatment of hyperuricemia and gout. Expert Opin Pharmacother. 2021.
[18] Suo Y, Fedor JG, et al. Molecular basis of the urate transporter URAT1 inhibition by gout drugs. Nat Commun. 2025.
[19] Data on file, Arthrosi Therapeutics. Clinical trial results of AR882.
[20] Hall J, et al. Pharmacokinetic-Pharmacodynamic Relationship of Verinurad, a Selective URAT1 Inhibitor, in Patients with Gout. Clin Pharmacol Drug Dev. 2020.
[21] Shiozawa A, et al. How URAT1 inhibitors can shape the future of chronic gout treatment: a narrative review of uricosurics past and present. Rheumatology (Oxford). 2021;60(10):4490-4498.
[22] Fitz-Patrick D, et al. Safety and efficacy of verinurad, a selective URAT1 inhibitor, for the treatment of patients with gout and/or asymptomatic hyperuricemia in the United States and Japan: Findings from two phase II trials. Mod Rheumatol. 2019.
李晓梅 撰写










